In der ICD-10 findet man nur unter dem Suchbegriff: Homocystin oder Homocystein (die Seiten von DIMDI und DIACOS kann ich nicht als Suchoption empfehlen) folgenden Eintrag:
- E72.1 (Störungen des Stoffwechsels schwefelhaltiger Aminosäuren)
Ich halte die u.g. Kodierung anfangs für ratsam, da es sich im screening zunächst nur serologisch nach- und beweisen läßt.
- R79.9 (sonstige näher bezeichnete abnorme Befunde der Blutchemie)
Bei dieser multifaktoriellen Entität spricht man von einer gestörten (Re)-Methylierung bzw. einem gestörten Homocyst(e)in-Metabolismus mit Erhöhung der Konzentration von Homocyst(e)in im Gewebe, Serum und Urin (ICD-10: R79.9) sowie den klinischen Folgen (ICD-10: E72.9).
Historie der Biochemie
- Newburgh stellte zwischen 1915-1925 die These auf, dass Proteine für die Bildung atheriosklerotischer Plaques verantwortlich sind.
- Müller isolierte 1922 eine „schwefelhaltige Aminosäure“.
- 1925 korrigierte Odake die Formel und gab ihr den Namen „Methionin“.
- 1931 bestimmten Barger und Coyne die Struktur von Methionin.
- 1932 beschrieb Vigneaud die chemische Struktur von Homocystein als Demethylierungsprodukt von Methionin.
- Ab 1939 synthetisierte Folkers Varianten des Vitamin B6 sowie mehrere Vitamin-B6-abhängige Enzyme.
- Die Reindarstellung der Folsäure (B9) gelang 1940–1948 Pfiffner, Stokstad und Snell.
- 1955 publizierte Hodgkin die Struktur von Cyanocobalamin (B12) per Röntgen-Strukturanalyse.
- 1959 beschrieb Field Symptome zweier Schwestern, bei denen 1962 Carson und Neil Cystein im Urin und Dent Homocystein im Urin fanden.
- Finkelstein und Mudd beschrieben 1965 den Methionin-Metabolismus.
- 1968 erforschte McCully genetischen Zusammenhang bei Homocystienurie durch Mangel an Cystathionin-β-Synthase mit dem Kofaktor B6.
- Später wurden weitere Defekte von Enzymen mit Vitaminen als Kofaktoren entdeckt, die ebenfalls zu Störungen im Methioninstoffwechsel führen können.
Abb.1 aus wikipedia.org
Grundlagen der Biochemie
- Homocystein (α-Amino-γ-Mercaptobutyrat) ist ein Zellgift und als L-Konfigurat ein Zwischenprodukt des C1- bzw. Ein-Kohlenstofftransfers. Es entsteht durch Methylierung der schwefelhaltigen Aminosäure L-Methionin über das Zwischenprodukt S-Adenosyl-Methionin (SAM), dem Methyl-Donator. Nach Demethylierung durch die Adenosyl-Homocysteinase entsteht S-Adenosyl-Homocystein (SAH), welches zu Homocystein hydrolysiert wird (Abb.2 aus [2]).Der Umbau erfolgt über die Remethylierung im Folsäure-Zyklus, welcher B12-abhängig ist. Neben der Methionin-Synthetase (MS) ist die Methylen-Tetra-Hydro-Folat-Reduktase (MTHFR) ein Schlüsselenzym. Der Abbau erfolgt alternativ und irreversibel über die B6-abhängige Kondensation mit Serin zu Cystein und Glutathion. Hier ist neben der Cystathionase die Cystathion-ß-Synthetase (CBS) das Schrittmacherenzym. [1] Homocystein wird zusätzlich in Leber und Niere über die Betain-Homocystein-Methyl-Transferase (BHMT) zu Methionin umgewandelt.
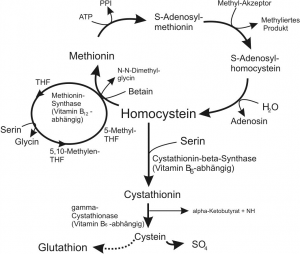
Abb.2 aus [1]
Ätiopathogenese
Die autosomal rezessiv vererbte klassische Homocysteinurie beruht auf einem Defekt der zytosolischen Cystathionin-ß-Synthase (CBS). Das Gen kann 60 verschiedene Mutationen aufweisen, wobei es auch heterozygote sowie Vitamin B6 beeinflussbare und nicht beeinflussbare Formen gibt. [2,3]
Der autosomal rezessiv vererbte Mangel an Aktivität der Methylen-Tetra-Hydro-Folat-Reduktase (MTHFR) beruht auf einem Gendefekt mit 25 Mutationsmöglichkeiten, wobei am häufigsten der Austausch von Alanin gegen Valin an der Position C677T vorkommt. Ein homozygoter (seltener heterozygoter) thermolabiler Genpolymorphismus verursacht regional unterschiedliche, mit bis zu 50% verminderte Leistungen der MTHFR. [3,4]
Einen weiteren Re-Methylierungsdefekt stellt die verminderte Aktivität der Methionin-Synthase (MS) dar. Er basiert auf einem Gendefekt oder einem zytosolischen Defekt der Synthese von Methylcobalamin und/oder Adenosylcobalamin.[3]
Es existieren vermutlich weitere Genpolymorphismen und –defekte mit Mangel an Vitaminen (v.a. der B-Gruppe). [2,3] Ein sekundärer Mangel an Vitamin B6, B9 und B12 entsteht durch Erkrankungen wie Adipositas, Hypertonie, Insulinresistenz und Hyperlipidämie [1], Nierenerkrankungen und Medikamente wie MTX [4], Alkoholkonsum [5], Rauchen, Kaffee und Bewegungsarmut. [6,7]
Ist der Abbau von Homocystein gestört, sind De- und Re-Methylierungsreaktionen in der Zelle gestört. [8] Genauere Zusammenhänge sind Gegenstand aktueller Trials. Leider wird Procain nur erwähnt, obwohl die demethylierende Effekte schon Ende der 80er Jahre bekannt, jedoch in vitro zu schwach bzw. in vivo nicht beweisen sind. [9,11]
Erhöhte Homocysteinspiegel resultieren neben den o.g. eingeschränkten Enzymaktivitäten (CBS, MTHFR, MS) und den erworbenen Hypovitamonosen auch aus der B12-abhängigen Methylmalonyl-Mutasen, weswegen hier auch eine Methylmalonacidämie möglich ist.
Es kommt dadurch zu einer toxischen Schädigung der Gefäßwand (Oxidation von Cholesterin und Lipoproteinen, erhöhte O2-Radikal- und erniedrigte NO-Bildung mit endothelialer Dysfunktion), erhöhter Thromboseneigung, Bildung von Schaumzellen, Plaques, Hyperplasie von glatten Muskelzellen im Plaque, Plättchenaggregation bzw. Hemmung von Thrombomodulin und Protein C. [2,12] Bekannt sind die Pathologien von Artheriosklerose, Herzinfarkt, Apoplex usw. aber auch Thrombose(neigung) und dadurch Erkrankungen wie Maculadegeneration, dementielle und MS-ähnliche Prozesse (durch Störungen des Stoffwechsels diverser Transmitter und des Myelin) mit Alzheimer und Depression sowie Osteoporose. [1,2,3,5,6,7,8]
differentialdiagnostische Einteilung
Neben der klassischen Homcysteinurie konzentriert man sich bei der Einteilung aufgrund der Höhe der serologischen Werte im Labor nach diversen Immunoassays in leichte, moderate und schwere Formen. Die DACH-LIGA sieht bei Werten < 10μmol/L keinen Handlungsbedarf, bei Werten von >10 – 12μmol/L nur einen Handlungsbedarf bei kardiovaskulärem Risikoprofil bzw. >12μmol/L eine Indikation zur Therapie. Bei seltenen Werten von >30 µmol/L besteht der Verdacht auf eine angeborene Störung (hetreo- bzw. homozygote Homocysteinämie bzw. -urie).
Therapie
Bei nachgewiesener Minderung der CBS kann und sollte man B6 bis max. 600mg/d bei gleichzeitiger Methionin-Diät <50mg/kg KG, bei Minderung der MTHFR nur Betain (jedoch 6-9g/d), bei Minderung der MS Cobalamine und Betain substituieren. [3]
Je nach Laborwert kann man bei leichten bis mittelschweren Fällen mit einer täglichen Gabe von B6=50mg, Folsäure=1mg und B12=1mg eine durchschnittlichen Senkung um 55% des erhöhten Homocysteinwertes erreichen. [10]
Bei angiologischen bzw. Gefäßerkrankungen kann Procain lokoregional sowie als Infusion über die Abbauprodukte Di-Ethyl-Amino-Ethanol (DEAE) und Para-Amino-Benzoe-Säure (PABA) helfen. [12]
Literatur
- Reuter ET (2010) Prädiktive Wertigkeit von Homocystein für die Mortalität von Dialysepatienten in einem unselektionierten Kollektiv eines tertiären Behandlungszentrums. Diss.
- Störk S, Angerer P, von Schacky C (1997) Hyperhomocysteinämie: ein neuer Risikofaktor für die Atherosklerose. Dtsch. Med. Wochenschr. 122:1-2
Harms E, Wendel U (2002) Störungen des Stoffwechsels von Aminosäuren und organischen Säuren. In Lentze MJ, Schaub J, Schulte FJ, Spranger J (Hrsg.) Pädiatrie. 2. überarb. und erw. Aufl. Springer
- Födinger M, Wölfl G, Fischer G, Rasoul-Rockenschaub S, Schmid R, Hörl WH (1999) Mutation (677 C to T) in the methylenetetrahydrofolate reductase gene aggravates hyperhomocysteinemia in hemodialysis patients. Kidney Int. 52(2): 517–23
- Bleich S, Carl M, Bayerlein K, Reulbach U, Biermann T, Hillemacher T, Bönsch D, Kornhuber J (2005) Evidence of increased homocysteine levels in alcoholism: the Franconian Alcoholism Research Studies (FARS). Alcohol Clin Exp Res.29: 334–6
- Ploder M, Schroecksnadel K, Spittler A, Neurauter G, Roth E, Fuchs D (2010) Moderate hyperhomocysteinemia in patients with multiple trauma and with sepsis predicts poor survival. Mol Med.
- Weikert C, Dierkes J, Hoffmann K, Berger K, Drogan D, Klipstein-Grobusch K, Spranger J, Möhlig M, Luley C, Boeing H (2007) B vitamin plasma levels and the risk of ischemic stroke and transient ischemic attack in a German cohort. Stroke 38(11):2912-8
- Durand P, Prost M, Loreau N (2001) Impaired homocysteine metabolism and atherothrombotic disease. Lab Invest 81: 645–72
- Hagemann S (2010) Charakterisierung von DNA-Demethylierungs-Mechanismen zur Optimierung der epigenetischen Therapie. Diss.
- Siegers CP, Budde U, Nägele H, Stubbe HM, Görne M (2013) Absenkung pathologisch erhöhter Homocysteinspiegel – Ergebnisse einer multizentrischen Dosisfindungsstudie zur Ermittlung eines optimal wirksamen Präparates zur Homocysteinsenkung.
- Villar-Garea A, Fraga MF, Espada J, Esteller M (2003) Procaine is a DNA-demethylating agent with growth-inhibitory effects in human cancer cells. Cancer research 63/16: 4984-9
Wander R, Gündling PW (2010). Naturheilverfahren bei Claudicatio intermittens. EHK 59(01): 5-12
